
|

|
|
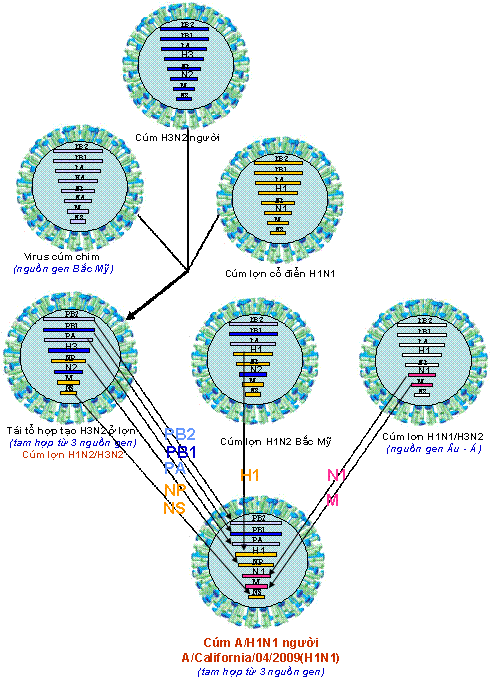
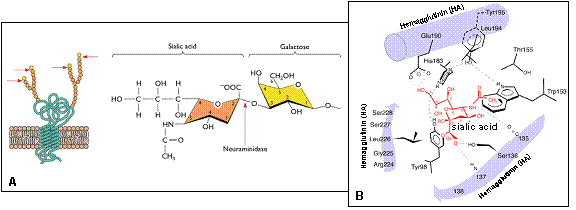
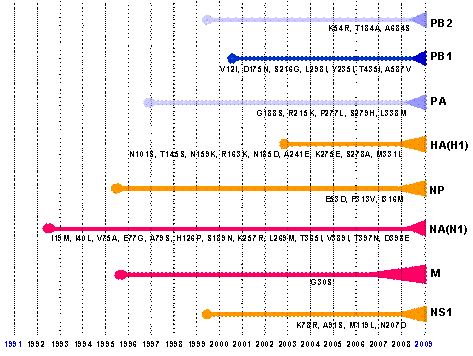
Virus cúm A (Influenza A virus) là một loại virus đặc biệt, có mức độ và điều kiện tiến hóa nhanh do khả năng thay đổi hệ gen tạo nên phân type và biến chủng mới thông qua cơ chế đột biến “lệch kháng nguyên” (antigenic drift) hoặc/và tái tổ hợp “trộn kháng nguyên” (antigenic shift). Trộn kháng nguyên là hiện tượng trao đổi các phân đoạn gen hay còn gọi là tái tổ hợp trao đổi (reassortment) giữa các virus thuộc cùng một phân type hoặc giữa các phân type khác nhau, dẫn đến sự hình thành một loại virus mới có đặc tính gây bệnh và kháng nguyên hoàn toàn khác có khả năng gây đại dịch toàn cầu. Virus cúm A/H1N1 – 2009 (A/(H1N1)v; S-OIV) hiện nay là điển hình của quá trình tiến hóa đặc biệt như vậy giữa các dòng virus cúm A. Một loạt virus cúm A gây đại dịch trong thế kỷ 20 và virus H1N1 hiện nay (2009) được hình thành là hệ quả của quá trình tiến hóa tái tổ hợp nhiều dòng của virus cúm ở động vật và người. Trong đó phải kể đến các dòng/phân type: i) H1N1 gây đại dịch cúm Tây Ban Nha (Spanish flu) năm 1918 – 1919; ii) H2N2 gây đại dịch cúm châu Á (Asian flu) năm 1957 – 1958; iii) H3N2 gây đại dịch cúm Hồng Kông (Hong Kong flu) năm 1968 – 1969; và iv) H1N1 mới hình thành, gây đại dịch cúm hiện nay, năm 2009. Dòng virus cúm A/H1N1 – 2009 có đặc tính di truyền, gây bệnh và kháng nguyên khác hẳn các dòng virus cúm A/H1N1 trước đó, đây là virus của người, được hình thành qua nhiều lớp tiến hóa từ nhiều nguồn gen và dòng gen của virus cúm A khác nhau. Đây là kết quả của sự tiến hóa “lệch kháng nguyên” và tái tổ hợp “trộn kháng nguyên” từ nhiều dòng virus của lợn, người và chim có xuất xứ Bắc Mỹ và Âu – Á. Gen polymerase PB2 và PA lấy từ nguồn gen cúm lợn H1N1/H3N2 mà trước đó tái tổ hợp từ cúm chim/lợn Bắc Mỹ; gen PB1 lấy từ cúm A/H3N2 của người; gen H1, NP và NS thu thập từ cúm lợn cổ điển H1N1 và cúm lợn H3N2; gen N1, M lấy từ cúm lợn H1N1/H3N2 nguồn gen Âu – Á. Tất cả các phân đoạn gen kiến tạo virus H1N1 – 2009 đã có những đột biến lớn so với nguồn gen tiền nhiễm, đặc biệt là gen HA(H1), có sai khác đến khoảng 28% về nucleotide và amino acid của HA1 và nhiều vị trí glycosyl hóa khác lạ so với HA của H1N1(1918) và H1N1(2008). Gen NA(N1) cũng có mức độ biến đổi tương tự. Việt Nam đang là một trong gần 150 quốc gia đang có đại dịch H1N1 – 2009, lây lan nhiều tỉnh/thành và con số nhiễm H1N1 – 2009 đang gia tăng nhanh chóng. Trong bài báo này chúng tôi trình bày những dữ liệu thông tin về nguồn gen và cơ chế tiến hóa phân tử của dòng/phân type cúm A/H1N1 – 2009 trở thành virus của người, cụ thể về sự xuất hiện, lưu hành; đặc điểm chung về tiến hóa; cơ chế của sự hình thành, quá trình biến đổi độc lực và di truyền; một số nét về can thiệp và phòng chống cần giải quyết. Sự xuất hiện và lưu hành cúm A/H1N1 – 2009 của người tại Việt Nam tạo thêm một đối mặt dịch tễ mới tại nước ta, song song với cúm A/H5N1 đã và đang hoành hành trong nước nhiều năm qua. Từ khóa: Cúm A, dòng gen, đại dịch, H1N1 2009, H2N2, H3N2,H5N1, nguồn gen, S-OIV, tiến hóa phân tử Từ tháng 4/2009 đến nay, một phân dòng mới của virus cúm A/H1N1 bùng nổ ở Mexico, nhanh chóng lan sang nhiều nước Bắc Mỹ, châu Âu và đến nay, gần 150 quốc gia trên thế giới, 134.500 người nhiễm, 816 người chết (đến 22/7/2009), trong đó có Việt Nam đã và đang lưu hành dịch cúm H1N1 mới này (Gallaher, 2009; IT, 2009; Kuntz-Simon, Madec, 2009; Neumann et al., 2009) (Hình 1). Có lẽ, vận chuyển hàng không đã nhanh chóng đưa virus phân phối khắp toàn cầu (Khan et al., 2009). Virus cúm mới, xuất hiện năm 2009, với tên viết tắt là A/H1N1 – 2009 hay A/(H1N1)v (variant), hoặc còn được gọi là virus cúm A có nguồn gốc từ lợn (swine-origin influenza A (H1N1) virus), ký hiệu S-OIV, được khẳng định một cách chắn chắn đó là H1N1 của người và người chính là vật chủ thích hợp cho sự truyền lây trực tiếp người – người (Naffakh, van der Werf, 2009; Michaelis et al., 2009). Do vận tốc, hình thức lây lan và mức độ gây bệnh của virus H1N1 –2009 trên phạm vi toàn cầu, ngày 11 tháng 6 năm 2009, Tổ chức Y tế thế giới (WHO) đã chính thức thông báo nâng lên cấp độ 6, cấp độ cao nhất và công bố đại dịch cúm A/H1N1 trên phạm vi toàn thế giới (WHO, 2009). Cúm A phân type H1N1 (A/H1N1) chưa rõ được hình thành từ bao giờ, mà chỉ được biết đến lần đầu tiên vào năm 1918 trong đại dịch cúm mang tính khốc liệt nhất trong lịch sử còn gọi là cúm Tây Ban Nha (Spanish flu), bởi quy mô dịch bệnh toàn cầu với trên 40 triệu người tử vong (Nelson et al., 2008; Vana et al., 2008; Investigation Team, 2009). Sau đại dịch đó, phân type cúm A/H1N1 tiếp tục lưu hành trong cộng đồng loài người trong giai đoạn 1918 – 1957, sau đó biến mất trong vòng 20 năm, rồi lại xuất hiện từ 1977 cho đến nay. Một trong những câu hỏi về tiến hóa chưa được giải đáp một cách triệt để đó là, bằng cách nào virus H1N1 độc lực cao nhất trong lịch sử được hình thành; và ở đâu, lúc nào, virus xuất hiện đầu tiên để rồi gây nên vụ dịch thảm khốc trong lịch sử loài người vào năm 1918 – 1919 (Taubenberger, Morens, 2006). Hình 1. Bản đồ phân bố đại dịch cúm A/H1N1 - 2009 trên phạm vi toàn cầu của Tổ chức Y tế thế giới Mặc dù theo ghi nhận, những năm 1847, 1889 cũng đã có nhiều vụ dịch có biểu hiện lâm sàng và quy mô tương tự như đại dịch cúm, nhưng liệu đó có phải là dịch cúm tiền nhiễm của đại dịch cúm A/H1N1 – 1918 hay không? Đến những năm 1930, khi những chủng virus cúm A thuộc phân type H1N1 được phân lập đầu tiên ở lợn, sau đó là ở người; và bằng phương pháp huyết thanh học người ta đã chứng minh chúng có mối liên quan kháng nguyên với virus cúm gây đại dịch năm 1918 (Shope, 1936; Zhou et al., 1999). Những năm cuối 1990, khi kỹ thuật giải trình tự được ứng dụng và những chuỗi gen đầu tiên của một số chủng virus đại dịch cúm cổ điển H1N1 – 1918 đã được giải mã, đó là những chuỗi gen thuộc về các chủng A/South Carolina/1/1918 (H1N1); chủng A/Brevig Mission/1/1918(H1N1) và chủng A/New York/1/1918(H1N1) (Taubenberger et al., 1997; 2005; Reid et al., 1999; 2000; 2002; 2004ª) cho phép có những hiểu biết phân tích liên quan sâu xa hơn giữa các chủng của phân type H1N1 tiền nhiễm và hậu nhiễm trong quần thể động vật và người (Fanning et al., 2002; Reid et al., 2003; Taubenberger et al., 2000; Vana et al., 2008; Zimmer, Burke, 2009). Những nghiên cứu tiếp tục sau đó cho thấy, sau năm 1918, virus cúm A/H1N1 vẫn tiếp tục lưu hành trong tự nhiên, bắt đầu phân hóa tạo thành các dòng lây nhiễm; hoặc có thể đã phân hóa trước đó, nhưng đến những năm 1918 – 1950 mới phát hiện thấy trong quần thể gia súc và người (Jordan, 1927; Oxford et al., 2002). Dòng H1N1 lưu hành trong quần thể loài lợn, có thể cũng vẫn song hành trong cộng đồng loài người, thực hiện quá trình tái tổ hợp đổi gen (antigenic shift) gây nên các ổ dịch hàng năm cho đến những năm 1950. Với sự xuất hiện của virus cúm H2N2 gây đại dịch trên người năm 1957 (còn gọi là đại dịch “cúm châu Á”), một hiện tượng thật lạ kỳ đã xảy ra là, những phân dòng trực tiếp của H1N1 từ dòng A/H1N1 – 1918 hoàn toàn biến mất khỏi cộng đồng loài người, nhưng “hậu duệ” của chúng vẫn cứ còn tồn tại trong quần thể loài lợn. Năm 1977, H1N1 của người lại xuất hiện, với nhiều bằng chứng cho rằng đó có thể là virus H1N1 của những năm 1950 lưu giữ trong phòng thí nghiệm bị thất thoát ra tự nhiên, do mức độ tương đồng gần như tuyệt đối giữa kháng nguyên các chủng sau 1977 với các chủng H1N1 gây bệnh trước đó 20 năm (Kendal et al., 1978). Các dòng virus này vẫn tiếp tục tồn tại trong tự nhiên, để cung cấp nguồn gen đa dòng kiến tạo nên các phân dòng virus cúm A mới (Taubenberger, Morens, 2006; Bahl et al., 2009). Năm 2006, hai dòng “hậu duệ” H1N1 của A/H1N1 – 1918 và hai dòng mới tái tổ hợp khác nguồn là dòng H1N1 của người và dòng H1N1 của lợn (cúm lợn ‘cổ điển”), đã tiến hành tái tổ hợp nhiều lần tạo nên dòng virus cúm A/H3N2 của người (Scholtissek et al., 1978; Zhou et al., 1999; Sun et al., 2009). Cũng từ dòng H3N2 này của người, cũng giống như sự phân hóa H1N1 trước đây, một dòng thích ứng H3N2 của lợn cũng được hình thành (Taubenberger, Morens, 2006; Yu et al., 2007; 2008; Sun et al., 2009). Mức độ gây bệnh đối với người của các phân type mới này không cao, bệnh xảy ra lẻ tẻ, cả hai dòng H1N1 và H3N2 của lợn hầu như không gây bệnh mà chỉ gây nhiễm đối với người (Olsen, 2002; Taubenberger, Morens, 2006). Từ đó trở đi, cúm A/H1N1 vẫn giữ mức độ độc lực gây nhiễm và gây bệnh “ôn hòa” trong quần thể lợn và cộng đồng người mãi cho đến tháng 4 năm 2009. Với nổ lực nghiên cứu vaccine cấp tốc, cuối tháng 5 năm 2009, Viện Tiêu chuẩn và Kiểm định Sinh phẩm Quốc gia (NIBSC) Vương quốc Anh đã tạo được giống virus vaccine chủng NIBRG-121 theo phương pháp di truyền ngược, lấy gen HA(H1) và NA(N1) từ chủng A/California/7/2009(H1N1)v; và ngày 27 tháng 5 năm 2009, Tổ chức Y tế thế giới công bố và khuyến cáo sử dụng chủng NIBRG-121 để sản xuất vaccine cho người phòng chống đại dịch H1N1 – 2009 hiện nay (http://www.who.int/csr/resources/publications/swineflu/2009_05_27IDCDC RG15a.pdf). Cùng với NIBSC, Trung tâm Kiểm soát và Phòng ngừa bệnh tật Hoa Kỳ (CDC) cũng đã thành công tạo giống vaccine H1N1 ký hiệu IDCDC-RG15; và một số giống vaccine khác CBER-RG2, X-179ª, IVR-153 phát triển ở các quốc gia khác cũng đã được WHO khuyến cáo sử dụng làmvaccine (http://www.who.int/csr/resources/publications/swineflu/en/). ĐẶC ĐIỂM CHUNG VỀ TIẾN HÓA PHÂN TỬ CỦA VIRUS CÚM A/H1N1 – 2009 Tái tổ hợp tạo nên virus cúm A/H1N1 – 2009 từ nguồn gen đa dòng Cúm A/H1N1 – 2009 đương nhiễm trên người hiện nay, có thể nói là kết quả của một quá trình tiến hóa hết sức đặc biệt để tạo nên một sản phẩm H1N1 qua nhiều lớp tiến hóa tái tổ hợp đa dòng (Shinde et al., 2009; Investigation Team, 2009; Gallaher, 2009). Giai đoạn đầu và giữa của thế kỷ 20, tính cách biệt địa lý và sự cạnh tranh loài vật chủ, đã tạo nên thế tiến hóa tách biệt của các lớp tổ hợp nguồn gen cúm A, hay còn gọi là bể gen (gene pool) theo hai lớp phân dòng: lớp phân dòng tổ tiên Âu – Á (Eurasian lineages) và lớp phân dòng tổ tiên châu Mỹ (American lineages) (Bahl et al., 2009). Những thập kỷ cuối thế kỷ 20 đầu thế kỷ 21, nguồn gen có cơ hội trộn nhập, mà một trong những loài vật chủ giao lưu nguồn gen chính là con người và phương tiện giao thông của con người (Khan et al., 2009; Naffakh, van der Werf, 2009). Cúm chim Bắc Mỹ có dịp tiếp xúc với cúm lợn Âu – Á; cúm người và cúm lợn cổ điển có dịp trao đổi gen cùng nhau để có cơ hội tái tổ hợp tạo nên các phân type và biến chủng mới, trong đó điển hình là H1N1 – 2009 (Zhou et al., 1999; Taubenberger, Morens, 2006; Bahl et al., 2009; Zimmer, Burke, 2009). Hình 2. Sơ đồ tiến hóa phân tử tái tổ hợp trao đổi (reassortment) tạo nên virus A/H1N1 – 2009 Cúm A/H1N1 – 2009 được hình thành sau hai quá trình tam hợp, đó là phân type của một tổ hợp các phân đoạn gen có mức độ biến đổi khác rất nhiều so với cúm A/H1N1 vốn vẫn đang lưu hành ở châu Mỹ trong các năm trước đó (cho đến 2008), đặc biệt đối với các gen kháng nguyên H1 và N1 (Gallaher, 2009; Shinde et al., 2009; Zimmer, Burke, 2009). Nguồn gốc tiến hóa tái tổ hợp tạo nên cúm A/H1N1 của người năm 2009 đặc trưng bằng sự trộn lẫn hỗn tạp của nhiều dòng gen từ cúm chim, người, lợn để tạo nên các chủng trung gian tái tổ hợp, cung cấp các phân đoạn gen cho sự hình thành cúm A/H1N1 ngày nay (Smith et al., 2009) (Bảng 1). Bảng 1. Lý lịch “trích ngang” của 8 phân đoạn gen cúm A/H1N1 A/California/04/2009(H1N1)).
Ghi chú: * PB1-F2 (273 bp): Lồng vào gen PB1 nhưng bị gián đoạn do đột biến; **M1 (759 bp); ***M2 (294 bp): hình thành do nối gen (splicing) trong phân đoạn M; ****NS1 (660 bp); *****NS2(NEP) (366 bp): hình thành do nối gen (splicing) trong phân đoạn NS. Lược tóm lịch sử hình thành các dòng cúm A cung cấp nguồn gen tiến hóa Dòng virus cúm A/H1N1 gây đại dịch 1918 – 1919 (cúm Tây Ban Nha) Đại dịch đầu tiên được ghi nhận trong lịch sử là cúm Tây Ban Nha do virus thuộc phân type H1N1 gây nên mà tính đặc hiệu kết hợp kháng nguyên của dòng virus H1N1 – 1918 này khác hoàn toàn với H1N1 của lợn lúc đó, cho phép nhận xét, dòng cúm A gây đại dịch trên người này đã tiến hóa và biến đổi (Shop, 1936). Đối với virus cúm A/H1N1 ở lợn, sau này được gọi là dòng virus “H1N1 cổ điển” của lợn (classical swine H1N1), cho đến khi phân lập vào năm 1930, có các đặc tính sinh học kháng nguyên và phân tử sau hơn 10 năm tiến hóa, đã khác xa nhiều so với H1N1 – 1918 của lợn năm 1918 và H1N1 – 1918 của người năm 1918 (Kanegae et al., 1994). Nhiều nhà nghiên cứu cho rằng, dòng virus cúm H1N1 – 1918 bắt nguồn từ Mỹ (vùng Kansas) và tràn sang châu Âu (Barry et al., 2008), nhưng cũng có nghiên cứu cho thấy, dòng virus H1N1 đầu tiên này vốn sinh ra ở các nước Đại Tây Dương ngay tháng 2 năm 1918 (Simonsen, 2004; Zimmer, Burke, 2009). Trước đó, năm 1916 – 1917, cũng đã có một số ổ dịch cúm xảy ra tại các căn cứ quân sự của Pháp, có lẽ, đây là nguồn virus tiền nhiễm để tiến hóa phát sinh đại dịch 1918. Cho dù còn tranh cãi thời gian nào là chính xác và ở đâu phát sinh nguồn gen đầu tiên của virus cúm A/H1N1 – 1918, nhưng thực tế hiển nhiên cho thấy đại dịch này không khởi phát tại châu Á, mặc dù sự góp nhặt nguồn gen kiến tạo vẫn còn là vấn đề bí ẩn cần làm sáng tỏ (Knobler et al., 2005). Đại dịch 1918 khởi phát bằng một đợt dịch “nhẹ” vào mùa xuân năm 1918 và “nặng” dần trong các tháng cuối 1918 đầu 1919. Cùng lúc, dịch phát ra ở châu Âu và Bắc Mỹ, chủ yếu là ở Mỹ tại các trung tâm huấn luyện và cơ sở quân sự của Mỹ chuẩn bị đưa quân sang châu Âu (Reid et al., 2001). Đợt dịch thứ hai tiếp nối vào mùa thu năm 1918; và trong gần 2 tháng, dịch bệnh đã lan toả khắp thế giới với tỷ lệ gây chết 5 – 10% (Knobler et al., 2005). Đợt thứ hai, do chịu ảnh hưởng của miễn dịch thu được từ đợt trước nên tỷ lệ được bảo vệ là 56 – 89% so với 35 – 94% trước đó, còn những đợt tiếp theo trong năm 1919 – 1920 dịch bệnh đã không còn mãnh liệt như trước (Barry et al., 2008). Cũng như các đại dịch cúm về sau, dịch cúm Tây Ban Nha (1918 – 1919) cũng bắt đầu bằng gây chết với tỷ lệ lớn do phá vỡ cân bằng miễn dịch bởi cơ chế của “cơn bão cytokine” (cytokine storm), rồi sau đó trong cộng đồng mặt bằng miễn dịch được nâng dần và sự mãnh liệt của dịch bệnh thuyên giảm dần rồi mất hẳn (Kobasa et al., 2007). Về đặc điểm di truyền học, dòng cúm A/H1N1 – 1918 trên cơ sở phân tích gen/hệ gen mới được giải mã gần đây cho thấy, khác với các chủng/dòng cúm gây đại dịch về sau này, các chủng phát sinh năm 1918 đều thực hiện quá trình kiến tạo hệ gen bắt nguồn từ cúm chim và chuyển đổi tính thích ứng sang người rồi lan truyền gây nhiễm trực tiếp người – người (Reid, Taubenberger, 1999; Taubenberger et al., 2005). Hệ gen của H1N1 – 1918 chắc chắn được lấy từ thành phần góp nhặt phân đoạn gen của cúm chim chưa biết rõ nguồn gốc phát sinh (Taubenberger, Morens, 2006; Vana, Westover, 2008). Các dòng virus cúm cổ điển và thường trực của lợn H1N1, H1N2, H3N1, H3N2 Sơ khai nhất, cúm A gây nhiễm trên lợn lần đầu tiên được ghi nhận lâm sàng là vào năm 1918, cùng lúc với đại dịch cúm trên người giai đoạn 1918 – 1919. Một điều cần phân biệt rõ ràng là, virus đại dịch cúm H1N1 – 1918 hình thành từ nguồn gen virus cúm chim mà không phải phát sinh từ nguồn gen đương nhiễm lúc đó đang có trên lợn (CIDRAP, 2009). Chủng cúm lợn H1N1 đầu tiên được phân lập vào năm 1930 ở Mỹ, từ đó cúm lợn H1N1 hầu như thường trực trong quần thể lợn ở mọi thời gian cho đến nay (Kanegae et al., 1994; Olsen, 2002), trở thành dòng virus cúm lợn cổ điển (classical swine strain/lineage). Dòng virus cúm lợn cổ điển H1N1 được phát hiện tồn tại gây bệnh đường hô hấp ở nhiều nước trên thế giới (Liu et al., 2009ª). Bên cạnh đó, còn có một số phân type khác tạo nên các dòng gây nhiễm cúm A trên lợn, bao gồm H1N2, H3N1 và H3N2, làm phức tạp hóa các dòng virus cúm A gây nhiễm và gây bệnh có nguồn gen tàng trữ trong lợn (Saito et al., 2008; Chutinimitkul et al., 2008; Moreno et al., 2009), tạo điều kiện cho sự trao đổi chéo hình thành phân type hoặc các chủng mới có độc lực cao hơn (Yu et al., 2007; 2008; Sun et al., 2009). Cúm lợn (swine influenza) được hiểu theo nghĩa dịch tễ học là dịch cúm gây ra do các chủng virus cúm A thông thường thích ứng và tồn tại thường xuyên trong vật chủ là lợn. Như đã nói, chủng nỗi trội gây dịch cúm ở lợn là thuộc phân type H1N1, một dòng virus cúm A tiếp tục tồn tại có thời gian xuất xứ từ những năm 1918, nhưng các chủng thuộc các phân type khác là H1N2, H3N1 và H3N2 cũng gây bệnh với triệu chứng bệnh tích tương tự và cũng được phân lập thường xuyên từ lợn (Taubenberger, Morens, 2006; Kothalawala et al., 2006; Ducatez et al., 2008; Michaelis et al., 2009). Cúm lợn xảy ra thường xuyên ở lợn tại nhiều nước trên thế giới với tỷ lệ nhiễm cao và tỷ lệ chết thấp (1 – 4%), trong đó có các tiểu bang miền Trung nước Mỹ, Mexico, Canada, Nam Mỹ, châu Âu (gồm Vương quốc Anh, Thuỵ Điển, Italy), Kenya, Trung Quốc, Đài Loan, Nhật Bản, Hàn Quốc, Thái Lan và một số nước khác (Olsen, 2002; Yu et al., 2007; 2008; Chutinimitkul et al., 2008; Kuntz-Simon, Madec, 2009). Lợn, một vật chủ có vai trò thiết yếu trong vòng tiến hóa của cúm A thích ứng gây bệnh trên và của người, có thể bị gây nhiễm bởi một loạt các phân type cúm A dịch cúm mùa vụ (seasonal influenza A viruses) của lợn, chim, người; trong đó dòng H3N2 vốn sở hữu của người cũng tồn tại thường xuyên ở lợn tạo nên một thế tái tổ hợp trộn gen hình thành các chủng mới (Kothalawala et al., 2006; Sun et al., 2009). Dòng H3N2 gây đại dịch ở người những năm 1968 (còn gọi là dịch cúm Hồng Kông) được coi là phát sinh từ lợn, sau khi giết chết gần 1 triệu người, lại “ôn hòa” trở lại và là virus cúm mùa vụ thường xuyên có mặt ở cộng đồng người và quần thể lợn (Yu et al., 2007; 2008), bắt buộc một số nước phải áp dụng chương trình vaccine theo mùa cho lợn và cho người (Baras et al., 2008). Các vụ dịch nỗi trội do cúm lợn gây ra ở người được ghi nhận năm 1976 và vaccine cúm lợn sử dụng cho người được tiến hành ở Mỹ cho gần 40 triệu người, đã đẩy lùi virus gây bệnh trên người, mặc dù có hiện tượng phản ứng vaccine tạo nên hội chứng Guillain-Barre(Sencer, Millar, 2006). Có thể tóm lược, các giai đoạn và quá trình hình thành các chủng/dòng/phân type cúm lợn như sau: - Từ 1930 đến 1998, cúm lợn vùng Bắc Mỹ chủ yếu do phân dòng cúm lợn cổ điển A/H1N1 gây ra. - Từ 1998, dòng virus cúm A/H3N2 với sự tái tổ hợp các phân đoạn gen từ các nguồn gen cúm A ở người, lợn, chim (quá trình tam hợp, triple reassortment) trở nên nỗi trội gây bệnh ở lợn vùng Bắc Mỹ, song hành cùng dòng virus cúm lợn cổ điển H1N1. - Cùng trong giai đoạn này, các chủng virus thuộc dòng mới H1N2, kết quả của tái tổ hợp trao đổi chéo giữa dòng trung gian H3N2 tam hợp trước đó với dòng virus cúm lợn cổ điển H1N1, cũng được phân lập tại Mỹ. - Dòng phân hóa khác là H3N1 cũng được phân lập trên lợn ở Mỹ trong 10 năm gần đây, có thể là kết quả tái tổ hợp của H3N2 của gà tây với H1N1 trên người (loại cổ điển, trước 2009) và các virus cúm A đương nhiễm trong quần thể lợn, trở nên một dòng mới góp tay cho tiến hóa tái tổ hợp trao đổi gen hình thành cúm A/H1N1 – 2009 (Lekcharoensuk et al., 2006; Michaelis et al., 2009; Peiris et al., 2009). - Dòng virus mới H1N1 – 2009 được phát hiện trên người từ tháng 4 năm 2009. Ngày 02/5/2009, Cơ quan giám sát thực phẩm Canada (CFIA, Canadian Food Inspection Agency) thông báo tìm thấy virus mới H1N1 – 2009 trên đàn lợn ở Alberta (CFIA, 2009), ví dụ đó là chủng A/swine/Alberta/OTH-33-24/2009(H1N1) (số đăng ký: GQ369425). Mới đầu, CFIA nghĩ rằng lợn bị nhiễm do tiếp xúc với người nuôi mang virus H1N1 – 2009, nhưng sau đó loại virus mới này được xác định là virus H1N1 – 2009 của lợn, nguồn lây chưa được rõ. Giữa tháng 6/2009, tương tự, loại virus mới H1N1 – 2009 của lợn cũng gây ra ổ dịch tại một trang trại ở Argentina theo thông báo của Tổ chức Thú y thế giới (OIE) với 30% có triệu chứng lâm sàng, không có tỷ lệ chết (OIE, 2009). Các chủng cúm A/H1N1 – 2009 trên lợn có thành phần gen HA(H1) và NA(N1) đồng nhất tới 99 - 100% với cúm A/H1N1 – 2009 của người. Sự phức tạp của quá trình tiến hóa phân dòng cúm A ở lợn, cộng thêm một điều kiện sinh học là lợn là loài vật chủ thích ứng chuyển đổi cấu trúc của virus cúm A lên người, cũng như đây là loài vật quá gần với cộng đồng người trong mối quan hệ lây nhiễm, đã tạo lợi thế hết sức thuận lợi cho việc hình thành các dòng mới gây đại dịch của virus cúm A. Các dòng virus cúm gây đại dịch ở người H2N2 (cúm châu Á) và H3N2 (cúm Hồng Kông) Cả hai dòng virus cúm A H2N2 và H3N2 này đều xuất phát tại châu Á, gây đại dịch cách nhau khoảng trên dưới 10 năm, trong giai đoạn 1957 – 1958 và 1968 – 1969. Đại dịch cúm châu Á (Asian flu) do dòng virus cúm A phân type H2N2 gây ra, được sinh ra nội tại tại Trung Quốc vào khoảng thời gian đầu năm 1957 (Scholtissek et al., 1978; Lindstrom et al., 2004). Virus H2N2 được phân lập tại Singapore tháng 2/1957 và Hồng Kông tháng 4/1957, nhưng đại dịch H2N2 lại xuất hiện ở Trung Quốc, Hồng Kông, tràn xuống phương Nam vào mùa hè 1957, sang Mỹ tháng 6/1957, gây chết khoảng 1 triệu người trong các năm 1957 – 1958 (Glezen, 1996). Dòng virus cúm A/H2N2 – 1957, tiếp nhận 3 phân đoạn gen có nguồn gốc thủy cầm, từ vịt hoang dã và 5 phân đoạn gen khác từ chủng virus cúm A đang lưu hành trên người lúc đó, trong đó gen HA(H2) được thu nhận từ vịt (Schäfer et al., 1993; Scholtissek et al., 1978; Lindstrom et al., 2004). Sau đó 10 năm, đại dịch cúm Hồng Kông xuất hiện với một dòng virus cúm A mới gây bệnh, đó là H3N2. Dòng virus gây đại dịch này thu nhận 2 phân đoạn gen mới, thông qua tái tổ hợp trao đổi gen (reassortment) từ nguồn tàng trữ vịt trời và giữ lại 6 phân đoạn gen có nguồn gốc từ virus cúm người vốn lưu hành lúc đó. Số người chết do H3N2 trong đại dịch này không nhiều, một phần do nguyên nhân là, tuy gen HA có thay đổi từ H2 sang H3 (theo cơ chế trộn gen tái tổ hợp), nhưng gen N2 vẫn giữ nguyên vẹn. Tuy N2 không phải là một kháng nguyên gây miễn dịch bảo vệ hoàn toàn khi virus xâm nhập, nhưng kháng thể do N2 sinh ra cũng góp phần trung hòa virus sau nhiễm và do vậy, làm biến đổi mức độ ác liệt của dịch cúm (Glezen, 1996; Scholtissek et al., 1978; Lindstrom et al., 2004). Một lý do khác, là trước đó trong lịch sử, phân đoạn gen H3 đã có tồn tại nên ít nhiều cũng có tạo nên mặt bằng miễn dịch ở một số người lớn tuổi, làm kìm hãm đáng kể mức độ và tốc độ lây truyền của virus H3N2 trong đại dịch những năm 1968 – 1969 (Simonsen et al., 2005). Cũng có thể, do áp lực như vậy, nên virus H3N2 tuy “ác liệt” lúc khởi đầu đại dịch, nhưng không lâu và trở nên “ôn hòa” tồn tại cùng con người để thỉnh thoảng gây “cúm mùa vụ”, đặc biệt ở các nước Bắc Mỹ từ đó cho đến nay (Baras et al., 2008). Tuy nhiên, điều đáng lo ngại là sự lưu giữ thường xuyên của một nguồn gen cúm A trong quần thể người, sẽ tạo điều kiện tái tổ hợp sinh ra những chủng virus mới; và chính xác điều đó đã xảy ra với dòng H1N1 – 2009 đương đại có sự góp phần đắc lực của dòng gen H3N2 của người này (Michaelis et al., 2009; Neumann et al., 2009; Smith et al., 2009; Zimmer, Burke, 2009). Cũng giống như sự song hành tồn tại của H1N1, các dòng H3N2 của người và H3N2 của lợn cũng đồng thời lưu hành trong cộng đồng loài người và quần thể loài lợn từ đó cho đến nay (Yu et al., 2008; Chutinimitkul et al., 2008;Sun et al., 2009). CƠ CHẾ CỦA QUÁ TRÌNH BIẾN ĐỔI DI TRUYỀN TẠO NÊN VIRUS CÚM A/H1N1 – 2009 CỦA NGƯỜI Biến đổi yếu tố độc lực gây nhiễm giữa các loài Virus cúm A đã được phân lập từ nhiều loài động vật, gia súc, gia cầm, thủy cầm, chim di cư và động vật hoang dã, chính chúng là nguồn tàng trữ nguồn gen cho quá trình tái tổ hợp kiến tạo các biến chủng gây nên tất cả dịch cúm A trên thế giới (Bahl et al., 2009). Động vật và người cũng chính là tập hợp vật chủ lưu trữ nguồn gen lây nhiễm giữa các loài với nhau, kể cả nguồn gen tạo nên virus cúm ở người (Horimoto, Kawaoka, 2001). Chi phối quá trình lây nhiễm giữa các loài là quy luật “rào cản loài sinh học” (species-barrier), mà theo đó, virus của loài này không hoặc khó gây nhiễm đối với vật chủ thuộc loài khác. Đối với cúm A, hạn chế thích ứng vật chủ vẫn tuân thủ theo cơ chế nói trên, tuy nhiên có phần lỏng lẽo hơn, do các phân đoạn của hệ gen virus cúm A có thể được vay mượn giữa các phân type của virus ở các loài khác nhau một cách dễ dàng (Horimoto, Kawaoka, 2001; Bahl et al., 2009). Virus cúm A của loài chim thường nhân lên rất kém ở người (Beare, Webster, 1991), còn virus cúm ở người lại không hoàn toàn thích ứng nhân lên ở thủy cầm (Vines et al., 1998; Ito et al., 2000). Một điều hết sức đáng quan tâm là ở chỗ, hầu như tất cả các biến chủng tái tổ hợp của virus cúm A đều chọn lợn (Sus scrofa) làm vật chủ thích ứng trung gian trong quá trình biến đổi hoặc/và chuyển đổi cơ chế thích ứng vật chủ cảm nhiễm của mình (Horimoto, Kawaoka, 2001). Không giống như loài lợn có vai trò tàng trữ, lưu giữ và cung ứng nguồn lây, các loại virus cúm A ở loài ngựa (Equus caballus) chưa thấy có bằng chứng xác thực truyền lây trực tiếp sang người (Medeiros et al., 2004). Rõ ràng, loài chim và loài lợn vẫn là các loài tiếp tay đắc lực cho việc chuyển đổi một biến chủng virus cúm A từ chỗ “thích ứng cảm nhiễm và gây nhiễm” trở thành chủng (hoặc phân type) virus “của” người và “gây bệnh” trên người (Homo sapiens). Quá trình này, đối với nhiều loại virus khác gây bệnh của người phải mất hàng trăm/nghìn năm, nhưng đối với cúm A, có thể chỉ được thực hiện trong vòng vài chục năm (Zhou et al., 1999; Olsen, 2002; Xu et al., 2004). Virus cúm A/H1N1 – 2009 của người xuất hiện năm 2009 hiện đang gây dịch tại 150 quốc gia trên thế giới, trong đó có Việt Nam, là một minh chứng hết sức xác thực của quá trình tiến hóa đó (Shinde et al., 2009; Liu et al., 2009b; Kuntz-Simon, Madec, 2009). Vậy, cơ chế chuyển đổi yếu tố độc lực trong quá trình tiến hóa tạo nên biến chủng mới hoặc phân type mới có độc lực đặc hiệu theo loài như thế nào? Đặc hiệu loài vật chủ (host specificity), khác với thích ứng vật chủ (host adaptivity) là ở chỗ thích ứng vật chủ chỉ giúp virus thích ứng gây nhiễm một thời gian mà không coi vật chủ đó là nguồn môi giới truyền lây trực tiếp; còn đặc hiệu loài vật chủ là một loại hình tương tác giữa virus cảm nhiễm và tế bào/cơ thể thích ứng hình thành do tiến hóa, mà ở đó, virus thực hiện được một cách hoàn toàn quá trình gây nhiễm và truyền lây trực tiếp giữa cá thể này với cá thể khác (Manrubia, Lazaro, 2006). Đối với cúm A và cúm A/H1N1, yếu tố độc lực quyết định rất lớn quá trình xâm nhiễm là mối liên kết và mức độ tương tác biểu hiện giữa protein hemagglutinin (HA) bề mặt của virus với thụ thể tế bào cảm nhiễm của vật chủ có cấu trúc từ sialic acid (SA) (Kumlin et al., 2008; Qi et al., 2009) (Hình 3). Hình 3. Thụ thể tế bào và liên kết sialic acid của thụ thể với hemagglutinin. A. Phân tử thụ thể bề mặt cắm gốc Virus cúm A/H1N1 – 2009 đã biến đổi các vị trí ở chuỗi polypeptide HA(H1) không còn giới hạn nhận biết sialic acid ở góc quay α2,3 mà hoàn toàn chỉ nhận biết sialic acid ở góc quay α2,6 ở thụ thể tế bào cảm nhiễm của người (Qi et al., 2009). Do vậy, người – người là chuỗi xích vật chủ hoàn toàn phù hợp của cúm A/H1N1 – 2009. Biến đổi gen thành phần của hệ gen cúm A/H1N1 – 2009 Đặc điểm biến đổi của gen hemagglutinin (HA(H1)) Phân đoạn gen quan trọng nhất trong 8 phân đoạn gen cấu trúc hệ gen của một virus cúm A là HA (hemagglutinin), một protein vỏ, mang tính kháng nguyên và do tham gia và tác động vào quá trình xâm nhiễm của virus, nên HA còn được coi là protein độc lực của virus (Kanegae et al., 1994; Gallaher et al., 2009). HA mang đặc tính kết hợp thụ thể với sialic acid có trên bề mặt tế bào nhiễm trong quá trình gây nhiễm và đặc tính gây ngưng kết khi tiếp xúc với bề mặt hồng cầu gà và ngựa, do vậy, HA dễ dàng sử dụng trong chẩn đoán sự có mặt của virus (Stevens et al., 2005; Kalbfuss et al., 2008; Qi et al., 2009). Điểm nhô ra ở đỉnh của protein HA tạo nên một “hốc lõm” để liên kết với thụ thể carbon hydrate qua cầu nối sialic acid của tế bào, và đây là vai trò thiết yếu, do vậy, trong nguyên lý tạo vaccine cần bảo đảm có kháng thể sinh ra trung hòa được hemagglutinin. Mức độ sinh ra của kháng thể và khả năng trung hòa kháng nguyên bề mặt HA của virus cúm A được lượng hóa bằng phương pháp chuẩn độ (titration) thông qua phản ứng ngăn trở hồng cầu (Kalbfuss et al., 2008). Về đặc tính kháng nguyên – miễn dịch tương quan với độc lực virus, trên phân tử protein HA có nhiều vị trí quyết định kháng nguyên (epitope) và vị trí bám dính thụ thể (receptor – binding site), mà ở đó, nếu có đột biến (thay đổi amino acid) hoặc/và gắn kết carbon hydrate, thì rất có thể, tính kháng nguyên và độc lực bị thay đổi tuỳ theo mức độ khác nhau (Schwarzer et al., 2009). Hình 4 trình bày so sánh đối chiếu trình tự amino acid protein hemagglutinin (HA(H1)) của virus cúm A/H1N1 phân lập trên người, bao gồm H1N1 – 2009 (virus đại dịch cúm 2009, một chủng châu Mỹ và một chủng châu Âu), virus H1N1 – 1918 và H1N1 – 2008. Hai chủng virus H1N1 mới phát sinh đại diện cho đại dịch cúm năm 2009 là A/New York/18/2009(H1N1), phân lập tại Mỹ (số đăng ký: FJ984355) và A/Paris/2591/2009(H1N1), phân lập tại Pháp (số đăng ký: GQ249333) được sử dụng song đôi, giữa chúng chỉ có sai khác amino acid tại vị trí 221 (T221S) và 240 (D240E), chứng tỏ H1N1 – 2009 gây đại dịch toàn cầu có mức độ tương đồng rất cao. Một chủng virus H1N1 gây đại dịch năm 1918 là A/South Carolina/1/18 (H1N1) (số đăng ký: AF117241); và một chủng virus H1N1 phân lập ở người năm 2008 là A/DistColumbia/WRAMC-1154048/2008(H1N1) tại Mỹ (số đăng ký:CY038770) được đưa vào so sánh với mục đích xác định mức độ không tương đồng giữa chủng gây đại dịch hiện tại (2009) với chủng H1N1 gây đại dịch cổ điển năm 1918 và với chủng H1N1 – 2008 trước khi đại dịch phát sinh (Hình 4). Sai khác amino acid khi so sánh song đôi được đánh dấu (X); đột biến mất một amino acid ở một số chủng được biểu hiện bằng dấu (-); amino acid arginine (R) của điểm cắt protease, giữa HA1 và HA2, cũng như amino acid glutamic acid (E) thiết yếu cho liên kết với sialic acid được đánh dấu như chỉ dẫn. Các amino acid tham gia vào 5 vùng epitope kháng nguyên cũng được đánh dấu chỉ dẫn bằng khung tên là vị trí A-E và các vị trí glycosyl hóa gồm 3 amino acid (N-X-S/T) được xác định để so sánh giữa các chủng. Gen HA(H1) có độ dài 1701 bp ở tất cả các chủng gây đại dịch 1918 và 2009; nhưng ở một số chủng H1N1 – 2008 gen này chỉ có kích thước 1698 bp. Vị trí cắt của protease ở giữa HA1 và HA2 chỉ có một amino acid kiềm là arginine (R), amino acid chịu trách nhiệm chính trong liên kết với sialic acid là glutamic acid (E) vẫn bảo tồn trong tất cả các chủng. Kết quả so sánh trình tự amino acid các chủng H1N1 – 2009 với H1N1 – 2008 cho thấy, 166 trên 566 amino acid có sai khác trong toàn bộ chuỗi polypeptide H1, chiếm tỷ lệ 18,73%; nếu chỉ so sánh 343 amino acid của HA1 thì có đến 93 amino acid sai khác, chiếm tỷ lệ 27,4%, còn trong chuỗi HA2, chỉ có 5,4% (12/223 amino acid) không tương đồng (Hình 4). Hình 4. So sánh đối chiếu trình tự amino acid protein hemagglutinin (HA(H1)) của virus cúm A/H1N1 năm 2009 Virus cúm A/H1N1 – 2009 của người thu nhậngen HA(H1) từ dòng gen H1N1 cúm lợn cổ điển có nguồn gốc Bắc Mỹ, trong đó gen H1 bắt đầu hình thành từ 2002-2003, từ đó đến nay gen H1 ở H1N1 – 2009 gây đại dịch đã có ít nhất 9 đột biến làm thay đổi amino acid tại các vị trí: N101S, T145S, N159K, R163K, N185D, A241E, K275E, S278A, M331L, với sự biến đổi thành phần sâu sắc trong năm 2008 – 2009 như đã phân tích ở trên (Hình 5). Như vậy, gen H1, một gen quyết định tính kháng nguyên và gây bệnh của của cúm A/H1N1 đã có biến đổi rất lớn ở các chủng gây đại dịch năm 2009, một vấn đề làm trở ngại quá trình sản xuất vaccine hiện nay. Hay nói cách khác, H1N1 – 2009 không hoặc một phần rất ít bị trung hòa bởi kháng thể kháng H1 của virus H1N1 – 2008 hoặc kháng thể do các virus H1N1 trước đó có trong tự nhiên hay do vaccine hình thành. Điều kiện bắt buộc để có được vaccine có hiệu lực là phải sử dụng nguồn gen H1 của các chủng gây đại dịch hiện tại (WHO, 2009). Hình 5. Nguồn gen, thời điểm hình thành dòng gen và mức độ đột biến của các gen thành phần Trên bề mặt virus cúm A còn có một loại protein vỏ mang đặc tính enzyme nhận biết và phá bỏ sialic acid của thụ thể tế bào nhiễm để giải phóng virus giúp hoàn thành chu kỳ nhân lên, đó là neuraminidase (NA) hay sialidase do phân đoạn 6 của hệ gen virus cúm A mã hóa (Air, Laver, 1989; Suzukiet al., 2005). Hemagglutinin (HA) trung gian liên kết thụ thể trong quá trình xâm nhập (giai đoạn vào trong tế bào), còn neuraminidase (NA) có trách nhiệm cắt liên kết khỏi thụ thể trong quá trình giải phóng virus (giai đoạn ra khỏi tế bào) để xâm nhập vào tế bào khác (Skehel et al., 2000; Matrosovich et al., 2004; 2006). Trung hòa protein NA với kháng thể đặc hiệu neuraminidase, hoặc vô hiệu hóa ngăn trở chức năng của enzyme neuraminidase của virus bằng các loại hóa dược (ví dụ: oseltamivir, biệt dược Tamiflu; zanamivir, biệt dược Relenza) sẽ kìm hãm số lượng virus không có cơ hội xâm nhập tiếp vào các tế bào khác để thực hiện quá trình lây nhiễm (Matrosovich et al., 2004; Bauer et al., 2009; Ekiert et al., 2009; Maurer-Stroh et al., 2009). NA, chiếm khoảng 20 – 25% tổng số protein bề mặt của virus cúm, được coi là protein kháng nguyên do vai trò kích thích miễn dịch và tham gia vào miễn dịch học nói chung của quá trình lây nhiễm virus cúm A, đồng thời cũng góp phần làm sạch mucin trên niêm mạc đường hô hấp giúp virus tiếp xúc thụ thể tế bào để gây nhiễm (Michaelis et al., 2009). Virus cúm A tuy ở các phân type khác nhưng có cùng type kháng nguyên NA, có thể góp phần làm giảm mức độ gây bệnh của virus đương nhiễm khác phân type HA, ví dụ, H2N2 vaccine có thể làm giảm đến 50% mức độ gây nhiễm của virus H3N2 do cùng chung nhau phân type N2 tạo miễn dịch chéo, mặc dù H3 và H2 có thể khác nhau (Ducatez et al., 2008). Nếu N1 của virus đại dịch H1N1 – 2009 có độ đồng nhất cao với các phân type N1 đương có trong các quần thể vật chủ (ví dụ: H1N1 cổ điển của lợn và người) thì miễn dịch vốn có từ H1N1 – 2008 (chủng cũ) sẽ góp phần làm thuyên giảm sự lan toả của H1N1 – 2009 (chủng mới). Rất tiếc, sự thể không như vậy, cũng giống như gen H1, gen N1 ở chủng mới gây đại dịch hiện nay đã có mức độ đột biến rất cao và là một thành phần gen hoàn toàn mới, khác biệt đến 18,2% so với gen N1 của virus H1N1 năm 2008 trên người (Michaelis et al., 2009). Gen N1 của H1N1 – 2009 gây đại dịch là một thành phần góp nhặt từ dòng gen cúm lợn Âu – Á, mà trước đó, được tái tổ hợp từ dòng gen cúm chim Âu – Á, với độ dài của gen là 1410 bp, không thay đổi kể từ năm 1918 (Bảng 1) (Maurer-Stroh et al., 2009). Nguồn gen NA này được kiến tạo từ những năm 1992 – 1993, cho đến nay, khi tham gia làm thành phần phân đoạn 6 của H1N1 – 2009, gen N1 hiện tại đã có 13 biến đổi cơ hữu, đó là ở các vị trí I19M, I40L, V75A, E77G, A79S, H126P, S189N, K257R, L269M, T365I, V389I, T397N, D398E (Hình 5). Khi phân tích phả hệ, kết quả cho thấy, gen N1 của tất cả các chủng H1N1 cho đến năm 2008 trên người, kể cả A/Puerto Rico/8/1934(H1N1), hay A/South Carolina/1/1918 (chủng gây đại dịch năm 1918) và các chủng của H1N1 của lợn đều tập trung cùng nhau trong cùng một nhóm, nghĩa là ít nhiều chúng cùng chia chung tính kháng nguyên; trong khi đó các chủng H1N1 – 2009 gây đại dịch hiện nay là hoàn toàn tách biệt (Babakir-Mina et al., 2009). Miễn dịch từ kháng nguyên N1 của các dòng H1N1-2008 hoặc của cúm lợn cổ điển để giúp trung hòa kháng nguyên N1 có trong H1N1 – 2009 là hoàn toàn không mong đợi được, do chênh lệch thành phần kháng nguyên quá lớn giữa các chủng (Michaelis et al., 2009; CIDRAP, 2009). Đặc điểm biến đổi của tổ hợp gen polymerase (PB2, PB1, PA) Virus cúm A/H1N1 – 2009 (S-OIV-H1N1) có 8 phân đoạn gen kiến tạo từ các dòng gen hỗn hợp mà trước đó nguồn gen cung cấp để thực hiện tái tổ hợp trao đổi được tiến hóa từ “bể gen” của virus cúm chim và lợn (Smith et al., 2009). Cụ thể, từ các dòng gen chính sau đây: từ dòng gen cổ điển của cúm lợn nguồn gốc Bắc Mỹ (gen hemagglutinin, HA; gen nucleoprotein, NP; gen non-structural protein, NS); từ dòng gen cúm chim nguồn gốc Bắc Mỹ (gen polymerase basic protein 2, PB2; gen polymerase acidic protein, PA); từ dòng gen cúm mùa vụ của người H3N2 (gen polymerase basic protein 1, PB1) và từ dòng gen cúm lợn cổ điển Âu – Á nguồn gốc từ cúm chim tại Âu – Á (gen neuraminidase, NA; gen matrix protein, MP) (Smith et al., 2009; Michaelis et al., 2009). Cụm gen PB2, PB1, PA có vai trò quyết định sự tổng hợp các thành phần của virus cúm A, trong đó có thành phần RNA kiến tạo hệ gen và các protein kiến tạo cấu trúc của virus (Beare, Webster, 1991; Taubenberger et al., 2005). Đối với virus H1N1 – 2009, tổ hợp gen polymerase lấy từ nguồn gen Bắc Mỹ của lợn và người, mà trước đó các dòng gen cung cấp chúng trong vòng 10 – 12 năm qua, được tiến hóa trải qua các giai đoạn tái tổ hợp (tam hợp, triple reassortment) (Bảng 1; Hình 5). Tổ hợp gen enzyme xúc tác tổng hợp này là kết quả của một sự hỗn hợp tiến hóa, từ 3 nguồn gen của 3 loại vật chủ: chim, người và lợn. Gen PB2 và PA có nguồn gốc từ cúm chim Bắc Mỹ, tái tổ hợp trung gian qua H3N2 của lợn, rồi bổ sung vào H1N1 – 2009; gen PB1 hoàn toàn lấy từ cúm H3N2 của người ở Bắc Mỹ, vốn lưu hành rộng rãi trong loài người (Hình 2). Trải qua 10 năm tiến hóa, bắt nguồn từ những năm 1998 – 1999, gen PB2 chỉ có 3 đột biến ở các vị trí K54R, T184A, A684S; trong khi đó gen PA cũng từ cúm chim được thu nhận từ những năm 1996 – 1997 lại có 5 đột biến bao gồm G188S, R215K, P277L, S279H, L338M; còn gen PB1, một gen đích thực của cúm người lấy từ H3N2 của người có hệ số đột biến cao nhất kể từ khi được hình thành vào những năm 2000 – 2001 đến nay, với 7 đột biến là V12I, D175N, S216G, L298I, Y235I, T435I, A587V (Hình 5). Nguồn gen polymerase của H1N1 – 2009 là của cúm chim và người, không có nguồn gốc phả hệ từ cúm lợn tuy có trải qua quá trình tái tổ hợp trung gian trên lợn (Smith et al., 2009; Neumann et al., 2009; Michaelis et al., 2009; Peiris et al., 2009). Lồng cùng chiều vào gen PB1 lệch khung đọc là gen PB1-F2, có độ dài 273 bp. Ở cúm A/H1N1 từ 1918 cho đến 2008, trên người và trên lợn, PB1-F2 mã hóa cho một loại protein nhỏ có chức năng gây chết tế bào theo chương trình (apoptosis) đối với các tế bào có nguồn gốc miễn dịch, đặc biệt là các đại thực bào phế nang (alveolar macrophage) theo cơ chế làm thay đổi hình thái, làm mất điện thế của màng ty thể (Coleman, 2007; Basler, Aguilar, 2008). Đối với virus gây đại dịch hiện nay (H1N1 – 2009 hay S-OIV), PB1-F2 không còn là một gen hoàn chỉnh nữa, do bị đột biến trong thành phần gen PB1 đã gây ảnh hưởng tiêu cực đến khung gen của PB1-F2 và chính sự “đột phá” về đột biến của PB1 để tăng tính độc lực đã “hy sinh” gen PB1-F2, mặc dù đối với hầu như tất cả virus cúm A cho đến nay, kể cả H5N1 gia cầm, PB1-F2 có vai trò không nhỏ trong hợp nhất biểu hiện độc lực của virus (Coleman, 2007). Đặc điểm biến đổi của các gen phụ trợ khác (MP, NP, NS) NS (non-structural protein) là gen mã hóa cho 2 loại protein bao gồm NS1 (660 bp) đa chức năng, có vai trò trong tăng cường độc lực và NS2 (hay còn gọi là NEP, nuclear export protein) độ dài 366 bp, hình thành do hiện tượng “nối gen” (splicing). Protein NS2 có nhiệm vụ kết hợp với protein đệm M1 (matrix protein 1) tương tác với yếu tố chuyển vận qua màng tế bào (CEF1, cellular export factor) để vận chuyển phức hợp vRNP (viral nucleoprotein) sau khi các phân đoạn hệ gen đã được bao gói trong protein NP (nucleoprotein) (Ye et al., 2006). MP hay còn gọi là M (matrix protein) kích thước 982 bp, bao gồm hai khung gen, một khung mã hóa cho M1 (759 bp); và khung kia cho M2 (294 bp), hình thành do hiện tượng “nối gen” (splicing). NP và NS là các phân đoạn gen đích thực của virus cúm lợn cổ điển H1N1 có nguồn gốc xuất xứ Bắc Mỹ (classical swine H1N1, North American lineage) mà trước đó những gen này đã được tái tổ hợp trong virus trung gian (Bảng 1; Hình 2) (Smith et al., 2009; Michaelis et al., 2009). NP có xuất xứ từ dòng gen hình thành vào những năm 1995 – 1996, trải qua 14 năm đã đột biến ở 3 vị trí amino acid là E53D, F313V, I316M; còn phân đoạn gen NS chỉ mới qua 10 năm tiến hóa, hình thành từ những năm 1999 – 2000, với 4 đột biến thay đổi amino acid tại các vị trí K78R, A91S, M119L, N207D (Hình 5). Phân đoạn gen MA tham gia vào hệ gen của H1N1 – 2009 từ dòng gen xa nhất, dòng gen cúm lợn H1N1/H3N2 xuất xứ Âu – Á, từ những năm 1995 – 1996, nhưng có vẻ là gen bảo tồn nhất trong 8 phân đoạn hệ gen, với duy nhất một biến đổi amino acid ở vị trí G30S (Hình 5). Rõ ràng, các phân đoạn gen NP, NS, MP chỉ mã hóa cho các loại protein có chức năng vận chuyển và bảo vệ của virus, nên mức độ tiến hóa có phần hạn chế và đều có nguồn gốc từ cúm lợn đích thực. MỘT SỐ VẤN ĐỀ VỀ CAN THIỆP VÀ PHÒNG CHỐNG Cúm A/H1N1 – 2009 với hóa dược điều trị Cho đến nay, hai nhóm hóa dược điều trị cúm A có giá trị và hiệu quả là: nhóm ngăn cản chức năng protein NA gồm oseltamivir (Tamiflu) và zanamivir (Relenza); và nhóm ngăn cản chức năng protein M2 là amantadine và rimantadine là các dẫn chất có nguồn gốc từ adamantane (Cinatl et al., 2007; Maurer-Stroh et al., 2009). Loại hóa dược oseltamivir và zanamivir ít độc, dễ sử dụng; và oseltamivir (Tamiflu) có lợi thế hơn vì sử dụng bằng đường uống thuận lợi hơn zanamivir (Relenza) bằng đường hít thở. Cho đến bây giờ, điều trị bệnh nhân nhiễm cúm A/H1N1 – 2009 với Tamiflu rất hiệu quả, chỉ có 1 trường hợp kháng thuốc xảy ra tại Đan Mạch (xem Michaelis et al., 2009). Đối với người lớn, uống 2 viên Tamiflu (viên nhộng 75 mg) trong một ngày, ngay khi có triệu chứng cúm. Sự phát triển kháng thuốc của virus H1N1 – 2009 với các loại hóa dược này chắc phải cần thời gian để đủ khả năng tiến hóa. - Chủng vaccine X-179ª: Là chủng tái tổ hợp được tạo ra bằng phương pháp gây nhiễm chéo, mang gen H1 (số đăng ký: GQ214335) và N1 (GQ214336), từ chủng đương nhiễm A/California/7/2009(H1N1)v, do Trường Đại học Y New York, Mỹ. -Chủng vaccine IDCDC-RG15: Là chủng tái tổ hợp lắp ráp gen H1 (GQ377047) và N1 (GQ377046) bằng kỹ thuật di truyền ngược, từ chủng đương nhiễm A/Texas/5/2009 (H1N1)v do Trung tâm hợp tác Giám sát và Phòng dịch của Tổ chức Y tế thế giới tại CDC (WHO Collaborating Centre for Surveillance, Epidemiology and Control of Influenza in the Centers for Disease Control and Prevention (CDC), Atlanta, GA, USA) tạo ra. - Chủng vaccine IVR-153: Cũng là chủng được tái tổ hợp do CSL, Australia tạo ra bằng kỹ thuật di truyền ngược, với gen H1 và N1 lấy từ chủng đương nhiễm A/California/04/2009 (H1N1)v, ngay lập tức được thử nghiệm trên người từ 29/6/2009 và dự kiến cung cấp khoảng 10 triệu liều/năm cho Australia. HƯỚNG TƯƠNG LAI VỀ TIẾN HÓA PHÂN TỬ Tiến hóa tái tổ hợp của virus cúm A nói chung và H1N1 nói riêng sẽ không bao giờ dừng lại (Taubenberger et al., 2001). Cho đến bây giờ, cúm H1N1 gây đại dịch hiện nay vẫn chưa biết do đâu và vì sao mà được phát sinh ra một cách đột ngột như vậy. Thực sự mà nói, cúm đại dịch hiện nay, không gây chết với tỷ lệ cao, nhưng gây nhiễm và lây lan nhanh với mức độ khinh khủng, đặt chúng ta vào sự cảnh giác giống như cúm H1N1 – 1918. Ban đầu cũng “nhẹ nhàng” như vậy, nhưng chỉ sau một thời gian ngắn cúm đại dịch H1N1 – 1918 đã “mạnh” lên rất nhanh, cướp đi gần 50 triệu sinh mạng. H1N1 – 1918 được coi là mẹ đẻ của các dòng cúm A hình thành sau những năm 1918 và là nguồn gốc cho các phân dòng gen đa dạng nhất tái tổ hợp tạo nên biến chủng (Reid et al., 2004b; Taubenberger, Morens, 2006). Sự xuất hiện hết sức đột ngột của một biến chủng virus gây đại dịch hiện nay là S-OIV (H1N1 – 2009; H1N1v), một dòng gen hoàn toàn mới, là minh chứng cho kết quả của một quá trình xảy ra nhanh và mạnh: một quá trình đột biến lệch gen gần như hoàn toàn của gen kháng nguyên H1 và N1; và một quá trình tái tổ hợp trao đổi các gen gần như hỗn hợp của các phân đoạn gen hỗ trợ khác (Gallaher, 2009; Michaelis et al., 2009; Zimmer SM, Burke, 2009; Neumann et al., 2009; Peiris et al., 2009). Một điều hết sức đáng lo ngại là sự hội nhập tiến hóa của các dòng gen cúm A, đã chọn nơi xảy ra trên vật chủ là lợn, một môi trường lý tưởng để chuyển gen và biến đổi gen. Dòng H1N1 – 2009 có thể chuyển biến theo 2 hướng: i) Thực hiện trao đổi gen chéo để tạo nên các biến chủng có mức độ độc lực cao hơn hiện nay, ví dụ, với H5N1 thể độc lực cao trên gia cầm xuất hiện trong 5 năm gần đây, rất có thể sẽ đồng hành cùng H1N1v (H1N1-2009) và biến đổi “giãn nở” (expanding) vị trí cắt của protease, từ một arginine (R) thành một loạt R và K (lysine) tăng độc lực; ii) Hoặc một kịch bản khác có thể xảy ra, đó là H1N1 – 2009 cũng sẽ thuyên giảm dần do mặt bằng miễn dịch trong cộng đồng ngày càng được tăng cường, do điều kiện vật chủ eo hẹp và virus H1N1 – 2009 sẽ được bình thường hóa như đã thấy với H3N2 gây cúm mùa vụ từ xưa đến nay. Có hai yếu tố tác động hỗ trợ cho virus H1N1 – 2009 tiến hóa theo xu hướng thứ 2, đó là: 1.Trong khi các virus đại dịch có độc lực cao trước đây H1N1 – 1918, H2N2 – 1957 và H3N2 – 1968, chỉ chấp nhận tái tổ hợp trao đổi phân đoạn gen HA(H1,H2,H3) và NA(N1,N2) đã biến đổi vào hệ gen của chúng, nhưng bảo tồn các phân đoạn khác vốn ít nhiều thích ứng với vật chủ là người (Taubenberger, Morens, 2006), thì H1N1 – 2009 gần như đổi một loạt tất cả các phân đoạn, phần nhiều lấy từ nguồn gen động vật (chim, lợn) (Gallaher, 2009). Điều này sẽ gây “khó dễ” cho virus gây đại dịch hiện tại trong vấn đề ổn định thích ứng xâm nhiễm có hiệu quả ở người; ii) Hơn nữa, hiện nay loại virus đại dịch mới này chưa kịp đối phó với các loại hóa dược ngăn cản neuraminidase (Tamiflu, Relenza), thêm vào đó miễn dịch mắc phải sẽ được hình thành, cùng với sự nỗ lực khống chế của thế giới kể cả áp dụng vaccine, sẽ “bắt ép” H1N1 – 2009 quay trở về quỹ đạo tiến hóa thông thường như các loại virus cúm mùa khác. Khác với cúm gây đại dịch trước đây, ngày nay thế giới đã có những mối liên kết dập dịch hiệu quả, với những phương tiện thông tin và công cụ hữu hiệu, mà một trong những biện pháp giá trị nhất vẫn là vaccine phòng dịch (WHO, 2009; Ortiz et al., 2009; Michaelis et al., 2009). Hàng chục tổ chức và công ty đang đầu tư nghiên cứu để có hàng chục chủng vaccine làm giống và khả năng sản xuất hàng tỷ liều vaccine đủ cung cấp cho nhu cầu thế giới là giải pháp khả thi được. Theo Tổ chức Y tế thế giới, từ tháng 10 hoặc 11 trở đi, có thể WHO khuyến cáo chương trình vaccine phòng chống H1N1 – 2009 qui mô thế giới. Việt Nam đã có kinh nghiệm đối phó với nhiều dịch bệnh, kể cả SARS, cúm A/H5N1, cùng với thế giới dưới sự hỗ trợ của WHO, nên chúng ta có quyền hy vọng kiểm soát được đại dịch này trên đất nước chúng ta.
GENETIC SOURCES AND MECHANISM OF MOLECULAR EVOLUTION OF THE A/H1N1 – 2009 INFLUENZA VIRUS CAUSING GLOBAL PANDEMIC IN HUMAN Institute of SUMMARY Influenza A virus is well known as a special entity of undergoing multiple evolutionary process to gain its capability for genetic changes to form novel lineage(s)/subtype(s) either through mutation, ‘antigen drift’ or reassortment, ‘antigen shift’. Antigen shift is a process of exchanging genetic materials derived from reassortment of gene segments between viruses, within and between subtypes, resulting in an antigenically novel virus that is capable of causing a worldwide pandemic. The novel A/H1N1 – 2009 (A/(H1N1)v; S-OIV) urrently causing global pandemic is a remarkable representative for such reassortment within and between lineages/subtype(s) of the influenza A viruses. A serial lineages and subtypes of influenza A causing pandemics in the 20th century and the present H1N1 (2009), are examples as consesquences of the evolutionary process of reassortment from many influenza A lineages/sublineages of human and animal origins. Among them, there have been encountered the lineages/subtypes: i) the H1N1 which caused the Spanish flu pandemic in 1918; ii) the H2N2 which caused the Asian flu pandemic in 1957 – 1958; iii) the H3N2 which caused the Hongkong flu pandemic in 1968 – 1969; and iv) the novel H1N1 reassortant which is causing the current 2009 pandemic. The A/H1N1 – 2009 influenza lineage genetically, antigentically and pathogenically differs completely from the previous H1N1 lineages; it is a human H1N1 to be formed through a number of evolution stages from many genetic sources and lineages of the influenza virus. This is the resulting reassortant of either antigen drift and antigen shift (reassortment) from many swine, human and avian lineages of North American and Eurasian origins. The polymerase PB2 and PA were obtained from the H3N2 swine genetic source which previously reassorted from the North American avian and swine influenza; the PB1 typically from the human A/H3N2; the hemagglutinin HA(H1), nucleoprotein NP and non-structural NS collected from the classical swine H1N1 and H3N2; the NA(N1) and M obtained from the Eurasian H1N1/H3N2 lineage of swine influenza. All the segments to construct the H1N1 – 2009 have undergone multiple genetic variations compared to the previous genetic sources, particularly, hemagglutinin HA(H1) differs about 28% in terms of nucleotide and amino acid of HA1 and different glycosylation sites from the HA of H1N1(1918) and H1N1(2008). Likewise, the NA(N1) of A/H1N1 – 2009 has such high level of variation. Key words: genetic source, H1N1 2009, H2N2, H3N2, H5N1, influenza A, lineage, molecular evolution, pandemic, S-OIV |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|



{kind=link}